Grant - Summer 2010 - DMD/BMD — Steve Wilton

MDA awarded a research grant totaling $368,100 to professor Steve Wilton at the University of Western Australia in Perth, for continued research into a strategy called "exon skipping," which bypasses mutations in the dystrophin gene responsible for Duchenne muscular dystrophy (DMD).
Wilton and his team were one of the first groups to use exon skipping to bypass DMD-causing mutations and partially restore production of the dystrophin protein missing in the disease. Since then, the exon-skipping field has rapidly progressed from an idea first demonstrated in studies of isolated cells to application in animal models of the disease and, more recently, to human clinical trials.
Two types of exon-skipping drugs that have reached the clinical trial stage are called 2Omethyl and morpholino. A third drug, 2 methoxyethyl, or "MOE," appears to be well-suited for exon skipping but was not made available for study until recently.
In their new work, Wilton and his team will conduct detailed preclinical studies in the mdx research mouse model of DMD to assess MOE's suitability for exon skipping.
In addition, the group has developed a number of other drugs designed to bypass a variety of different dystrophin mutations that should be responsive to exon skipping. The investigators will study the effectiveness and side-effect profiles of the new drugs.
"Without MDA support, the exon-skipping field would not be anywhere near as advanced as it is," Wilton said. "MDA funded this research when many other research funding bodies were very skeptical."
Funding for this MDA grant began August 1, 2010.
Grantee: DMD/BMD — Steve Wilton
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2011 - DMD/BMD — Tejvir Khurana, M.D., Ph.D.

MDA has awarded a research grant totaling $379,500 over three years to Tejvir Khurana, a professor of physiology at the University of Pennsylvania. The grant will support Khurana's research to increase the production of utrophin, a protein that may improve muscle strength and function in Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD).
Studies suggest that utrophin can, at least in part, substitute for the dystrophin protein missing in DMD and only partially functional in BMD. A mutation in the dystrophin gene is the root cause of both diseases.
Since functional utrophin is produced in the muscles of people with DMD and BMD, therapies based on supplying more of it are unlikely to cause an unwanted immune response. In contrast, therapies based on supplying functional dystrophin, which many people with DMD/BMD do not produce, may cause an unwanted immune response to this protein.
Khurana's strategy for increasing utrophin production is based on removing a biological "brake" that represses it.
"We have found that utrophin is in a state of repression," Khurana said, "and that a class of molecules called microRNAs cause this repression. We will develop methods to 'repress the repressors' and hence achieve utrophin upregulation."
Khurana and colleagues will test their strategy in dystrophin-deficient mice that develop a DMD-like disease.
Funding for this MDA grant began August 1, 2011.
Grantee: DMD/BMD — Tejvir Khurana, M.D., Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - DMD/BMD — Sean Forbes

MDA awarded $179,327 to scientific researcher Sean Forbes at the University of Florida in Gainesville for research into impaired blood flow to muscles lacking the dystrophin protein in Duchenne muscular dystrophy (DMD).
Forbes plans to study an enzyme called neuronal nitric oxide synthase (nNOS) that contributes to blood flow and delivery of oxygen to muscles in the mdx research mouse model of DMD and another mouse model lacking nNOS. He’ll test the hypothesis that impaired blood flow during and following muscle use leads to inadequate delivery of oxygen to the muscles in DMD, contributing to muscle damage and accelerating the loss of healthy muscle tissue.
Forbes and his team will attempt to improve muscle blood flow via administration of the drug sildenafil citrate (brand name Viagra) and observe whether it lessens damage in DMD muscles. The group will use magnetic resonance imaging (MRI) and another type of imaging called spectroscopy (MRS) to measure changes in muscle blood flow, energy production and damage in mice. These noninvasive techniques may prove to be sensitive tools to monitor and visualize disease progression and effectiveness of treatment in DMD.
"This MDA Development Grant provides a valuable opportunity to pursue this exciting and important area of research. The funding will enable us to perform studies to examine muscle metabolism and blood flow in dystrophy, and will examine the effects of a prospective treatment for DMD," Forbes said. "These studies have the potential to set the foundation for future studies in children with DMD."
Funding for this MDA grant began August 1, 2010.
Grantee: DMD/BMD — Sean Forbes
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2011 - DMD/BMD — Peter Currie, Ph.D.

Peter Currie, a professor of medicine at Monash University in Clayton, Victoria, Australia, was awarded an MDA research grant totaling $375,000 over three years. The funds will help support Currie's research, which involves screening for possible therapeutic molecules using the zebrafish model of Duchenne muscular dystrophy (DMD).
The underlying cause of DMD is an absence of the muscle protein dystrophin due to any of a large number of mutations in the dystrophin gene. Becker muscular dystrophy (BMD), a less severe form of MD, is caused by dystrophin gene mutations that result in production of a partially functional dystrophin protein.
"While the current animal models of DMD have generated valuable insights into the pathological basis of the disease, each has their limitations," Currie notes. "We have isolated mutations in the zebrafish dystrophin gene and have determined that dystrophin-deficient zebrafish accurately model the human condition."
In this new project, Currie and his colleagues will undertake a drug screen for small molecules that can inhibit the onset of signs of muscular dystrophy in the dystrophin-deficient zebrafish.
"MDA funding has been critical in allowing me to pursue modeling muscular dystrophy in zebrafish," Currie said. "There is simply no way that the research that has happened around muscle disease modeling in my laboratory would have occurred at the pace that it has without the generous support of MDA."
Funding for this MDA grant began August 1, 2011.
Grantee: DMD/BMD — Peter Currie, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - DMD/BMD — Michele Calos
Michele Calos, professor in the department of genetics at Stanford University School of Medicine in Stanford, Calif., received a $200,000 MDA research grant to develop a new stem cell-based therapy for Duchenne muscular dystrophy (DMD).
"Funding from MDA is critical for us to develop these new ideas that could lead to novel strategies to combat DMD," Calos said. "A prior MDA grant has allowed us to make progress in the studies, and we are now poised to perform the critical experiments."
The new work involves extraction of stem cells from DMD mice. A good copy of the dystrophin gene, necessary but missing in DMD, will be added to the cells, which will then be returned to the mouse where they are expected to home in on damaged muscle and help create new muscle fibers to replace those damaged by the disease. The investigative team will also conduct experiments using a type of adult human stem cell called "adipose-derived mesenchymal stem cells," which are purified from fat tissue and have been shown to develop into muscle.
Successful results in these experiments eventually may lead to clinical trials and may indicate potential for use of a similar approach in other muscle diseases.
"Progress in the development of stem cell therapies offers major hope for regeneration of muscle mass and strength," Calos said, even for those in whom the disease has progressed.
Funding for this MDA grant began August 1, 2010.
Grantee: DMD/BMD — Michele Calos
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2011 - DMD/BMD — Paul Martin, Ph.D.

Paul Martin, a professor of pediatrics and of physiology and cell biology at Ohio State University College of Medicine, has been awarded an MDA grant totaling $396,000 over three years. The award will help support Martin's study of the possible therapeutic effects of an enzyme called GALGT2 in Duchenne muscular dystrophy (DMD).
GALGT2 is involved in adding sugar molecules to a protein called alpha-dystroglycan, part of a cluster of proteins located at the muscle-fiber membrane. This cluster is abnormal in a number of forms of muscular dystrophy.
Among the other proteins normally located in this cluster are dystrophin, which is deficient in DMD and abnormal in the related Becker muscular dystrophy (BMD); alpha-sarcoglycan, which is deficient in the type 2D form of limb-girdle muscular dystrophy (LGMD); and merosin, which is deficient in the type 1A form of congenital muscular dystrophy (CMD).
Previous work in Martin's lab has shown that raising GALGT2 levels can help compensate for the loss of dystrophin in a mouse model of DMD, inhibiting the development of muscle abnormalities in DMD model mice.
The Martin lab also has shown that increasing GALGT2 levels can have therapeutic effects on skeletal muscles in animal models of LGMD2D and CMD1A.
"Because GALGT2 is normally expressed in skeletal muscles," Martin said, "its overexpression may have fewer immunologic issues than gene replacement or exon skipping approaches." (The latter approaches increase levels of proteins that are abnormal or deficient in patients, potentially triggering an unwanted immune response.)
"MDA has played a seminal role in the development of my work," Martin said. "MDA funded two years of my postdoctoral training, provided me with my first research grant as an independent faculty member, and now will provide research funding to translate proof-of-concept studies into potential therapies."
Funding for this MDA grant began August 1, 2011.
Grantee: DMD/BMD — Paul Martin, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - DMD/BMD — Louis Kunkel

MDA awarded a grant totaling $375,000 to Louis Kunkel, director of the program in genomics at Children's Hospital in Boston, for research into compounds already approved for human use that may alter disease progression in Duchenne muscular dystrophy (DMD).
"There are thousands of compounds developed for use in humans for a variety purposes such as pain, diabetes and heart disease," Kunkel said. "The vast majority of these compounds have never been tested for their possible use in other diseases such as muscular dystrophy, something that can be easily accomplished in zebra fish."
In previous studies, Kunkel's lab has identified several compounds that increase the survival rate in zebra fish with a DMD-like disease and that are approved by the U.S. Food and Drug Administration for use in humans.
The team now plans to search for additional compounds, which it will test in the zebra fish model. The investigators will analyze indicators including survival rates, muscle function and toxicity, to determine the best candidate to go forward into mouse models.
"The beauty of the zebra fish is that they are permeable," Kunkel said. "They present an opportunity to directly test compounds in a live organism with the disease due to their large numbers, small size, transparency and their ability to absorb chemicals from their water environment."
Funding for this MDA grant began August 1, 2010.
Grantee: DMD/BMD — Louis Kunkel
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2011 - DMD/BMD — Michael Rudnicki, Ph.D.

MDA has awarded a grant totaling $375,000 over three years to Michael Rudnicki, director of the Regenerative Medicine Program at Ottawa Hospital Research Institute and a professor in the department of medicine at the University of Ottawa, Canada. This funding will support Rudnicki's continuing studies on the function of muscle satellite cells in mice with a disease resembling human Duchenne muscular dystrophy (DMD).
DMD is caused by any of a number of mutations in the gene for the muscle protein dystrophin that result in a complete absence of the dystrophin protein in the skeletal muscles and heart. The closely related disease Becker muscular dystrophy (BMD) is due to mutations in the dystrophin gene that result in dystrophin protein that's only partially functional.
Muscle satellite cells are required for the growth and repair of skeletal muscles. In previous studies, Rudnicki and his colleagues have found that there is a subset of muscle satellite cells that function as "satellite stem cells," giving rise to the satellite cells that then become muscle.
They've also found that satellite stem cells do not function properly in dystrophin-deficient mice; and that a protein called WNT7a stimulates proliferation of satellite stem cells and benefits normal and dystrophic muscle.
In this new set of experiments, Rudnicki's team will investigate the nature of the defect in satellite stem cells in dystrophin-deficient mice and the mechanism through which WNT7a treatment increases the numbers of these cells.
The researchers also will investigate the possibility of using WNT7a as a treatment for DMD or BMD, through experiments in dystrophin-deficient mice.
Rudnicki, who has received MDA support for several years, says, "MDA funding has been critical in allowing my lab to investigate the fundamental mechanisms that regulate the function of muscle stem cells."
Funding for this MDA grant began August 1, 2011.
Grantee: DMD/BMD — Michael Rudnicki, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - DMD/BMD — Josephine Nalbantoglu
Josephine Nalbantoglu, associate professor in the department of neurology and neurosurgery at McGill University in Montreal, Canada, received an MDA grant totaling $313,170 for research into increasing levels of the muscle protein utrophin as a therapeutic strategy in Duchenne muscular dystrophy (DMD).
Utrophin is structurally similar to the crucial muscle protein dystrophin, missing in DMD. Nalbantoglu and colleagues aim to validate, in muscle cells and in an animal model of DMD, the potential for utrophin to fulfill some of the functions normally carried out by dystrophin and compensate for the missing protein.
In completed studies, Nalbantoglu's group has shown in an animal model of DMD that utrophin levels can be increased by targeting specific sites in the gene that carries the instructions for utrophin with proteins called transcription factors. Muscle injected with these proteins exhibited fewer hallmarks of disease and increased function despite the absence of dystrophin.
Using the same approach, the team will now refine transcription factors designed to result in increased levels of the human utrophin protein. In parallel, the investigators will explore various methods for delivering the factors throughout the body.
"Funding from MDA is crucial for this work," Nalbantoglu said. The approach of using artificial transcription factors to increase levels of specific genes would have wide application to other muscle diseases. Artificial transcription factors are already in clinical trials for some non-muscle diseases."
Funding for this MDA grant began August 1, 2010.
Grantee: DMD/BMD — Josephine Nalbantoglu
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2011 - DMD/BMD — Melissa Spencer, Ph.D.
Melissa Spencer, professor of neurology and co-director of the Center for Duchenne Muscular Dystrophy at the University of California, Los Angeles, has been awarded an MDA grant totaling $501,493 over three years. The funds will help support Spencer's continuing studies of the role of a protein called osteopontin and of inflammatory processes in a mouse model of Duchenne muscular dystrophy (DMD).
DMD results from a nearly complete absence of the dystrophin protein in the skeletal muscles and heart. Becker muscular dystrophy (BMD), a closely related disease, results from a partially functional dystrophin protein. A large variety of mutations in the dystrophin gene give rise to DMD or BMD.
The osteopontin protein has been shown to play a role in promoting the entry of immune system cells into damaged, dystrophin-deficient muscle, which in turn results in scar-tissue formation (fibrosis).
The Spencer lab has received funding from MDA over the past decade. Spencer and her colleagues, particularly Irina Kramerova and Carrie Miceli, will now study how osteopontin regulates the muscle-invading immune system cells and the manner in which these cells cause fibrosis in mice with a DMD-like disease.
The investigators will use mouse models and cellular and immunological techniques to understand the processes involved. They also will use a specialized mouse that has been genetically engineered to allow for noninvasive imaging of muscles.
These studies will lay the foundation for development of drugs that target osteopontin in DMD or BMD, Spencer said.
"I credit MDA for enabling many of the successes my lab has achieved," she noted.
Funding for this MDA grant began August 1, 2011.
Grantee: DMD/BMD — Melissa Spencer, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - DMD/BMD — Gordon Lynch

MDA awarded $375,000 to Gordon Lynch, professor of physiology at the University of Melbourne, Australia, for research into strategies aimed at improving muscle function in Duchenne muscular dystrophy (DMD).
Lynch's research builds on previous studies conducted by his group and others that have demonstrated the potential of proteins called "growth factors" to preserve muscle fibers. These growth factors are regulated by a family of proteins called insulin-like growth factor binding proteins (IGFBPs).
Lynch's team has uncovered evidence that a particular IGF binding protein called IGFBP-2 may play a key role in the cycles of muscle fiber damage and repair that occur in DMD.
Using a mouse model genetically altered to exhibit either increased or decreased IGFBP-2 activity, the team will seek to determine whether the protein aggravates or improves the DMD disease process. The researchers also will study the effects of IGFBP-2 and other IGF binding proteins on muscle development and muscle repair.
"The MDA has generously supported my laboratory’s research for over a decade, allowing my team to make consistent and significant contributions to the field, including demonstrating the exciting potential of growth factors for improving muscle function in muscular dystrophy," Lynch said. "Support from the MDA has been critical in facilitating my mentorship of some very talented young doctoral students and postdoctoral scientists who have each made important contributions to muscular dystrophy research and become successful independent investigators in their own right."
Lynch's new research will generate important new information about the functional roles of IGF binding proteins in muscular dystrophy, and contribute to the development of therapies designed to improve the quality of life for those with muscle disease.
Funding for this MDA grant began August 1, 2010.
Grantee: DMD/BMD — Gordon Lynch
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2011 - DMD/BMD — Martin Childers, Ph.D.
Martin Childers, a professor at the Institute for Regenerative Medicine in Winston-Salem, N.C., has been awarded an MDA grant totaling $480,000 over three years. The Institute is part of Wake Forest University Baptist Medical Center.
The new funds will help support Childers' research on heart disease associated with Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD). DMD results from a complete lack of dystrophin protein in skeletal and cardiac muscles, and BMD results from the presence of partially functional dystrophin protein. Both diseases are caused by mutations in the dystrophin gene.
"Many patients with DMD die as young adults from heart failure," Childers said. "The reason for this is that the heart muscle carries a genetic mutation that damages the normal operation of this all-important muscle. Our project will allow for the discovery of new drugs that might reverse or prevent the effects of the disease on the heart muscle."
Childers and colleagues will use two types of new technology to find potential new drugs for heart disease related to dystrophin deficiency. First, they'll use a method called cellular "reprogramming" to create heart cells from the skin cells of DMD patients. "This remarkable new technology will allow us to study how a genetic mutation affects the heart cells of a specific patient," Childers said.
The second method the investigators will employ is a drug-discovery "platform" that can screen thousands of compounds in individual cells.
"By marrying these two incredible technologies, this project will allow, for the first time, the testing of new drugs directly on the heart cells of an individual patient without any risk to the patient," Childers noted.
Funding for this MDA grant began August 1, 2011.
Grantee: DMD/BMD — Martin Childers, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - DMD/BMD — Dawn Cornelison

MDA awarded a grant totaling $160,000 to Dawn Cornelison, assistant professor in the division of biology at the University of Missouri-Columbia, for research into the role of a protein called syndecan-4 in muscle tissue repair after damage.
After muscle injury, Cornelison said, satellite cells (immature muscle cells) move in to make repairs. In order to do their job, the satellite cells need to perform a series of activities: They must first be 'activated' by signals resulting from injury; then divide several times to make more potential muscle cells; then stop dividing; and finally mature, or differentiate, into functional muscle.
In previous MDA-supported work, Cornelison and her research team analyzed a mouse engineered to lack the syndecan-4 protein and found that its satellite cells were unable to perform all of their normal muscle-repair functions. The group found that different parts of the syndecan-4 protein are responsible for regulating different satellite-cell functions in the muscle-repair process.
In its new work the group aims to identify proteins that interact with syndecan-4 and gain a better understanding of how the satellite cells carry out the four activities necessary for muscle repair.
"Funding from MDA has been critical to the progress and development of both this project and my scientific career," Cornelison said. "Particularly in the current environment, with the odds of obtaining funding for general research falling below 10 percent, knowing that MDA is there to sponsor research into muscle disease and regeneration is critical to retaining scientists in the field and encouraging others to undertake research into muscle diseases."
Funding for this MDA grant began August 1, 2010.
Grantee: DMD/BMD — Dawn Cornelison
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2011 - DMD/BMD — Emanuela Gussoni, Ph.D.
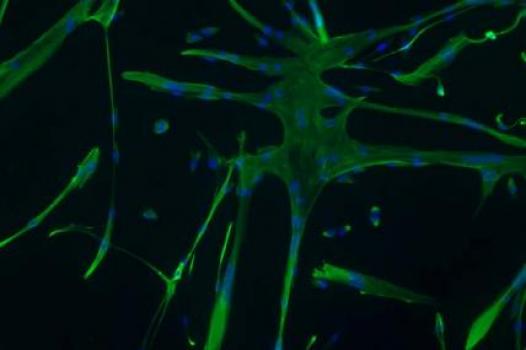
MDA has awarded a grant totaling $384,066 over three years to Emanuela Gussoni, an assistant professor in the division of genetics and program in genomics at Children's Hospital Boston and Harvard University. The grant will support Gussoni's studies to improve the efficacy of stem cell transplantation as a strategy to treat Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD) and possibly other forms of muscular dystrophy.
The direct injection of normal muscle stem cells into mice and people with muscular dystrophy has been attempted as a way to provide missing proteins to muscle tissue. (In DMD, the dystrophin protein is missing; in BMD, it's present but only partially functional.)
However, Gussoni said, studies have shown that, while the technique is safe in patients, their muscles have not shown significant improvement. One of the causes for the lack of improvement is the rapid death of the normal cells following transplantation.
To overcome this problem, different cell types more capable of surviving the transplant and producing the desired proteins must be identified. Gussoni's research team has found that human cells that produce a surface protein called MCAM can form muscle in tissue culture and following injection into animals.
If they live up to the preliminary expectations of Gussoni and her colleagues, MCAM-expressing cells could become candidates for cell transplantation to treat MD.
"MDA funding has been instrumental throughout my career," said Gussoni, who has received several MDA grants. "MDA develops and fosters both new and established investigators who are highly committed to improving the lives of people with muscle disorders."
Funding for this MDA grant began August 1, 2011.
Grantee: DMD/BMD — Emanuela Gussoni, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - DMD/BMD — Carmen Bertoni
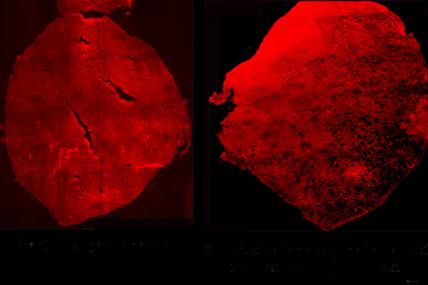
MDA awarded $408,915 to Carmen Bertoni, assistant professor of neurology at the University of California, Los Angeles, to continue research into DNA repair strategies in Duchenne muscular dystrophy (DMD).
Bertoni's research group has pioneered a technology that uses small molecules called single-stranded oligonucleotides, or ssODNs, to act directly on the source of the problem in DMD: the "genetic blueprint," or DNA. In people with DMD, the DNA that makes up the dystrophin gene contains errors that lead to missing or nonfunctional dystrophin protein.
"We use ssODNs to let the muscle know of those errors and give the opportunity to each cell that composes muscles to correct the mistake," Bertoni said. The fix is permanent, negating the need for ongoing treatment.
The process involves injection of the oligonucleotide into muscle, where it seeks the flawed region of DNA and highlights the specific error. The cell is alerted to the presence of the genetic defect, and activates the repair process to correct the flaw.
To date, the main limitation of ssODN-mediated gene correction has been the low frequency of gene repair. Bertoni's group aims to increase the strategy's efficiency to generate levels of gene repair that are clinically meaningful as a viable treatment for DMD.
The group will test the oligonucleotides first in muscle cells and then, once investigators have optimized the structure, in dystrophin deficient mice.
"MDA has had and continues to have a pivotal role in the development of this technology for the treatment of DMD," Bertoni said. "Without any question, the advance of clinical applications to DMD using ssODNs heavily relies on the help that continues to be received by the MDA. It is a great honor to be part of the numerous and extremely talented scientists that are supported by the Association."
Funding for this MDA grant began August 1, 2010.
Grantee: DMD/BMD — Carmen Bertoni
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - DMD/BMD — Bernard Jasmin

MDA awarded Professor Bernard Jasmin, vice-dean of research at the University of Ottawa in Canada, a grant totaling $360,000 to study the impact of small molecules called "exercise mimetics" in Duchenne muscular dystrophy (DMD).
One approach to counteract the effects of the loss of the muscle protein dystrophin in DMD involves increasing levels of a similar structural protein called utrophin. Studies performed in a widely used mouse model of DMD, the mdx mouse, have shown that increased utrophin expression can compensate for the lack of dystrophin and ameliorate symptoms of the disease.
Because utrophin activity normally is low and focused only in certain regions of the muscle, Jasmin's laboratory looks for ways to increase levels of the protein in DMD muscles.
"Our first preclinical study showed that treatment of mdx mice with an exercise mimetic known as GW501516 resulted in an increase in utrophin expression, improved muscle function and improved pathological symptoms of the disease," Jasmin said.
His current work builds on the previous study results, with plans for continued testing of GW501516 in the mdx mouse model of DMD, and expansion of the research to include testing of other molecules including AICAR, beta-GPA and resveratrol.
Because of the possible effects of exercise mimetics on utrophin in DMD-affected muscles, the small molecules hold potential for dramatic improvement in quality of life for individuals with the disease. GW501516 currently is under development to treat obesity by the pharmaceutical company GlaxoSmithKline, which could help speed its development for therapeutic use in people with DMD.
"MDA funding has allowed us to take a relatively basic molecular observation and perform research that eventually could result in development of novel therapeutic approaches for treatment of people with DMD," Jasmin said.
Funding for this MDA grant began August 1, 2010.
Grantee: DMD/BMD — Bernard Jasmin
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2011 - DMD/BMD — Alessandra Sacco, Ph.D.

MDA has awarded a research grant totaling $447,092 over three years to Alessandra Sacco, an assistant professor in the Muscle Development and Regeneration Program at Sanford-Burnham Medical Research Institute in La Jolla, Calif. The grant will help support Sacco's research on early-stage transplantation of muscle stem cells to treat a mouse model of Duchenne muscular dystrophy (DMD).
The goal of Sacco's new study is to understand whether transplanting muscle stem cells into a DMD mouse model during the fetal or newborn period can prevent the muscle degenerative process seen in DMD.
Sacco said that, by the time DMD is diagnosed and treatment begins in children with the disease, significant damage to muscles has accumulated, which "poses a major hurdle to the design of efficient therapeutic approaches."
She said transplantation of muscle stem cells during fetal or neonatal life may enhance the regenerative potential of skeletal muscles, resulting in an improved therapeutic outcome.
Sacco's group will make use of a recently developed DMD mouse model, in which the mice lack two proteins. Like people with DMD, they lack dystrophin, a muscle protein that's completely absent in this disease and only partially functional in people with the related Becker muscular dystrophy (BMD); and they also lack telomerase, a repair enzyme. These mice have a severe DMD-like disease.
If the transplantation approach is successful in these mice, it could have applications to human DMD, BMD and perhaps other forms of MD.
"MDA has been fundamental to my career development," Sacco said. "During my postdoctoral training, I received an MDA development grant for studying the contribution of bone-marrow-derived cells to skeletal-muscle regeneration. In the difficult funding climate of the last few years, the efforts of this organization to promote research in this field have been outstanding."
Funding for this MDA grant began August 1, 2011.
Grantee: DMD/BMD — Alessandra Sacco, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - DMD/BMD — Ashok Kumar

MDA awarded associate professor Ashok Kumar at the University of Louisville School of Medicine in Louisville, Ky., a grant totaling $349,206 for continued study of the molecular mechanisms that underlie disease onset and progression in Duchenne muscular dystrophy (DMD).
Kumar's team will work to identify biochemical changes in skeletal muscle caused by the loss of functional dystrophin protein in the disease.
"Recent studies in my laboratory have identified a set of proteins known as 'matrix metalloproteinase' (MMPs) that are highly deregulated in dystrophic muscles," Kumar said. "Interestingly, MMPs are the major enzymes responsible for tissue degradation in a number of disease states and conditions."
Kumar and colleagues have found that treatment with a molecule called batimastat is highly effective at ameliorating disease symptoms such as fiber necrosis (death) and inflammation, and improving skeletal muscle structure in dystrophin-deficient mice.
Now, the team will investigate the therapeutic potential of MMP-inhibiting drugs, some of which are already being used for other diseases in humans (including chemical relatives of the antibiotic tetracycline).
"I strongly believe that the current funding provided by MDA will open new avenues for treatment of DMD in the near future," Kumar said. "I sincerely thank MDA for its generous funding to pursue my research work in DMD."
Funding for this MDA grant began August 1, 2010.
Grantee: DMD/BMD — Ashok Kumar
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - CMT — Albena Jordanova
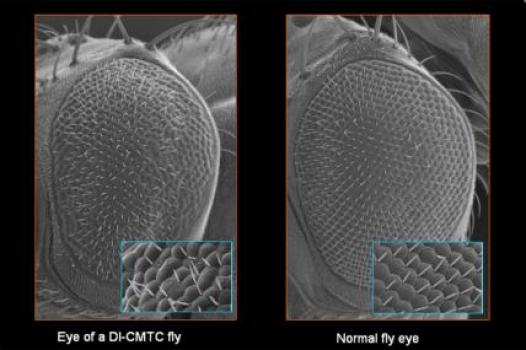
MDA awarded a grant totaling $282,630 to Albena Jordanova, professor in the department of genetics at the University of Antwerp, Belgium, for research into the molecular causes of, and potential drug targets for, a recently discovered form of Charcot-Marie-Tooth (CMT) disease known as dominant intermediate CMT type C (DI-CMTC).
Jordanova's research team was the first to describe the new CMT type and showed that the disease is caused by different mutations (genetic flaws) in the gene that carries instructions for an enzyme called tyrosyl-tRNA synthetase (YARS).
In their new work, Jordanova and colleagues aim to identify genes that modify the neurodegenerative symptoms of DI-CMTC present in a fruit fly research model of the disease. The team will then test the modifying genes to determine how well each is able to interact with drug-like chemical compounds, in an effort to pinpoint the best drug targets for DI-CMT and other types of the disease.
The "best gene hits" will be those that carry instructions for proteins that are both structurally favorable — that is, their protein folds are amenable to interactions with drug-like chemical components — and functionally favorable with a proven ability to affect the DI-CMTC disease process.
"MDA funding is absolutely essential to start this work," Jordanova said. "This is important seeding money for us to initiate a midterm research program and to perform the research we dream of."
Funding for this MDA grant began August 1, 2010.
Grantee: CMT — Albena Jordanova
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2011 – DMD - David Goldhamer, Ph.D.

MDA has awarded a research grant totaling $375,000 over three years to David Goldhamer, associate professor, director of the Center for Regenerative Biology, and associate director of the UConn Stem Cell Institute at the University of Connecticut in Storrs. The new funds will help support Goldhamer’s study of muscle stem cells and the repair of damaged muscle in Duchenne muscular dystrophy (DMD).
"Satellite" cells are adult muscle stem cells that are responsible for postnatal muscle growth and the repair of damaged muscle in injury and disease. In DMD, accumulation of intramuscular fat and connective tissue represent hallmarks of advanced disease, and the presence of these non-muscle tissues significantly affects muscle structure and function.
Satellite cells have been implicated as a possible cell of origin for increased fat and fibrotic (scarred) tissue in DMD muscle, but their involvement remains uncertain and controversial.
Using a research mouse model with a DMD-like disease, Goldhamer and colleagues will examine whether satellite cells promote increased fat and fibrosis in diseased muscle. In addition, the team will study regulators of muscle regeneration that may interact with or regulate satellite cell functions.
Results from Goldhamer’s work may help determine the potential benefits of modulating satellite cell behavior as a therapeutic strategy in DMD.
Funding for this MDA grant began February 1, 2011.
Grantee: DMD - David Goldhamer, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2011 - DMD — Margaret Zacharin, M.D.

Margaret Zacharin, an associate professor in the department of endocrinology and diabetes at Royal Children's Hospital in Parkville, Victoria, Australia, has been awarded an MDA research grant totaling $268,021 over two years. The award will help support a clinical trial of the drug zoledronic acid in children and adolescents with Duchenne muscular dystrophy (DMD).
Zoledronic acid is part of a class of medications known as bisphosphonates, which are used to prevent bone loss in osteoporosis and other bone conditions.
Children with DMD lose bone in part because of their underlying disease and in part because of the effects of corticosteroid medications. These are commonly used to prolong muscle function in DMD but have adverse effects on bone density.
"Bisphosphonates alter the course of corticosteroid-induced bone loss and largely prevent this complication in the adult population," Zacharin said. "It's more difficult to provide such evidence in a pediatric population." However, she noted, with newer techniques of calculation available, more accurate data can now be obtained.
"We aim to determine whether intravenous zoledronic acid plus vitamin D and calcium is better than vitamin D and calcium alone in increasing spinal bone density, reducing bone turnover and/or reducing fracture rate over two years in boys with Duchenne muscular dystrophy."
Zacharin will work closely with Craig Munns, an associate professor in the Department of Endocrinology at Children’s Hospital in Westmead, New South Wales, Australia, on this project.
She said results of the zoledronic-acid study may be used to develop a larger trial to assess fracture reduction in the DMD population and might ultimately be extended to other diseases, including other neuromuscular disorders.
MDA support, she said, "will allow us to undertake and complete this important study in a timely fashion."
Funding for this MDA grant began August 1, 2011.
Grantee: DMD — Margaret Zacharin, M.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - CMS — Michael Francis

Michael Francis, associate professor in the department of neurobiology at the University of Massachusetts Medical School in Worcester, received an MDA grant totaling $330,000 for research into effects on the connection between nerve and muscle known as the neuromuscular junction, or NMJ, in congenital myasthenic syndrome (CMS).
Francis and colleagues have engineered a research model using C. elegans, a type of worm also known as a nematode, that mimics many of the key features of a form of CMS called slow-channel congenital myasthenic syndrome (SCCMS). The team will use sophisticated genetic tools paired with measurements of the electrical properties such as voltage changes and electric currents in cells and tissues, and a sophisticated imaging technique called confocal microscopy in its analysis of NMJ function in both normal and diseased tissues.
The investigators plan also to pinpoint the molecular mechanisms underlying degeneration and alterations in NMJ function caused by neuromuscular disorders.
"We expect our efforts will provide key mechanistic insights into conserved molecular pathways important for NMJ development and function in the normal organism, as well as during diseases that impact NMJ function, such as SCCMS," Francis said. "Identifying the components of these pathways will be important for the development of new therapeutic strategies to combat neuromuscular disorders."
Funding for this MDA grant began August 1, 2010.
Grantee: CMS — Michael Francis
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2011 - DMD — Jasprina Noordermeer, Ph.D.

MDA has awarded a research grant totaling $278,570 over three years to Jasprina Noordermeer, a professor in the department of molecular cell biology at Leiden University Medical Center in the Netherlands. The grant will support Noordermeer's studies of the role of dystrophin in the brain. The dystrophin protein is absent or deficient in boys with Duchenne muscular dystrophy (DMD).
The role of dystrophin in skeletal muscles is fairly well-understood, and its absence in DMD leads to the progressive weakness seen in this disease. A great deal is also known about the role of this protein in cardiac muscle, where its absence leads to cardiac muscle abnormalities (cardiomyopathy) in DMD.
It's also known that dystrophin is normally present in the brain, and that at least some DMD patients have cognitive disabilities. However, little is known about the role of dystrophin or the effects of dystrophin deficiency in the brain.
Ultimately, Noordermeer says, treatments for DMD should reverse both muscle loss and the cognitive deficits associated with this disease, but how the lack of dystrophin results in defects at the connections ("synapses") where nerve cells interact with each other is unclear.
She and her colleagues will study the role of dystrophin and dystrophin deficiency in the brain in dystrophin-deficient flies and mice.
"MDA funding is critical to allow us to further our understanding of the functions of dystrophin and its [molecular] partners at the synapse," Noordermeer said. "We particularly appreciate MDA's commitment to funding research in model organisms directed toward unraveling DMD-associated dysfunctions."
Funding for this MDA grant began August 1, 2011.
Grantee: DMD — Jasprina Noordermeer, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
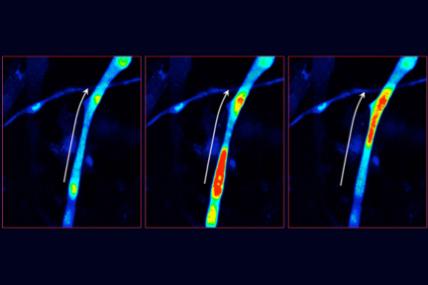
Grant - Summer 2010 - CCD — Francesco Muntoni
MDA awarded a grant totaling $375,000 to Francesco Muntoni, professor of pediatric neurology at University College London (UCL), United Kingdom, for research into the molecular mechanisms underlying central core disease (CCD) and multiminicore myopathies. Muntoni and Michael Duchen, professor of physiology, and cell and developmental biology, also at UCL, will work together, focusing on the mechanisms that lead to muscle weakness in the two diseases.
CCD and multiminicore myopathies both are caused by mutations in the ryanodine receptor gene (RYR1), which carries instructions for the RYR1 protein.
"While we have known for a number of years that RYR1 gene mutations were associated with these core myopathies, the precise mechanism leading to muscle weakness in affected individuals is less clear," Muntoni said.
It's known that RYR1 plays a key role in the release of calcium in skeletal muscle; the calcium release is followed by muscle contraction.
Muntoni and Duchen will investigate the hypothesis that chronic calcium dysregulation due to dysfunctional RYR1 protein leads to malfunction, in each muscle fiber, of the cell's energy factories, called mitochondria. With muscle cells from individuals carrying RYR1 mutations, the two will study various aspects of mitochondria health and function following exposure to abnormal calcium release.
"This study is a follow-on of a currently funded MDA grant that is coming to an end," Muntoni said. "The current MDA grant allowed us to improve our insight on the correlation between different mutations in RYR1 and the core myopathies. Without the original MDA grant, we would not have been in a position to establish these novel correlations between RYR1 and congenital diseases."
An improved understanding of the link between dysregulated calcium handling and mitochondria dysfunction could lead to new therapeutic interventions for individuals with core myopathies.
Funding for this MDA grant began August 1, 2010.
Grantee: CCD — Francesco Muntoni
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2011 – DMD - Bradley Olwin, Ph.D.
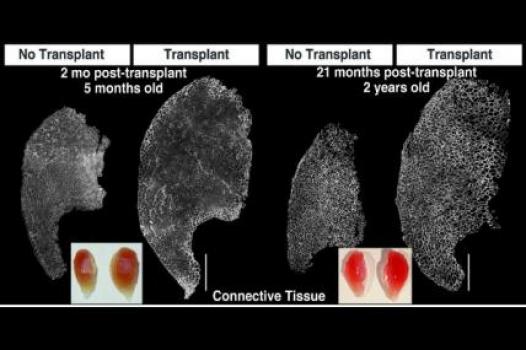
MDA has awarded a research grant totaling $369,165 over three years to Bradley Olwin, professor of molecular, cellular & developmental biology at the University of Colorado in Boulder. The new funds will help support Olwin’s study of muscle regeneration in injured and diseased skeletal muscle — particularly in the muscular dystrophies, including Duchenne muscular dystrophy (DMD).
Olwin is a longtime MDA grantee, having received funding from the Association almost continuously since the early 1980s.
Diseases such as the muscular dystrophies that result in progressive loss of skeletal muscle tissue reflect disruption of the normal muscle regenerative processes. If the cells responsible for normal repair could be augmented by drug therapies or cell replacement therapies, regeneration might be enhanced, slowing or halting the loss of skeletal muscle function.
In previous studies, Olwin and colleagues identified a rare stem cell that they hypothesize is a primary source of skeletal muscle stem cells (also called "satellite" cells). The team plans to study the newly identified cells and uncover any contributions they may make to the regeneration process in injured and diseased skeletal muscle. Next, the team will transplant the cells into the muscles of a research mouse with a DMD-like disease in order to determine how well they restore muscle function, slow muscle scarring (called fibrosis), and produce the protein missing in DMD, dystrophin.
Findings from Olwin’s studies may result in new drug-based or cell-based strategies to enhance new muscle growth in progressive muscle diseases.
"I have been funded by the MDA for the majority of my career as a research scientist, beginning in the early 1980s," Olwin said. "Without MDA funding, I may not have continued in skeletal muscle research."
Funding for this MDA grant began February 1, 2011.
Grantee: DMD - Bradley Olwin, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.

