Metabolic Myopathies
Types of Metabolic Myopathies
This section provides information about 11 metabolic diseases of muscle. Each one gets its name from the substance that’s lacking.
For more information about these types, please click on the individual disease name.
- Acid Maltase Deficiency (AMD, Pompe disease, glycogenosis type 2, lysosomal storage disease)
- Carnitine deficiency
- Carnitine Palmityl Transferase deficiency (CPT deficiency)
- Debrancher Enzyme deficiency (Cori or Forbes disease, glycogenosis type 3)
- Lactate Dehydrogenase deficiency (glycogenosis type 11)
- Myoadenylate deaminase deficiency
- Phosphofructokinase deficiency (Tarui disease, glycogenosis type 7)
- Phosphogylcerate Kinase deficiency (glycogenosis type 9)
- Phosphogylcerate Mutase deficiency (glycogenosis type 10)
- Phosphorylase deficiency (McArdle disease, myophosphorylase deficiency, glycogenosis type 5)
- Adenylosuccinate Synthetase 1 (ADSS1) Myopathy
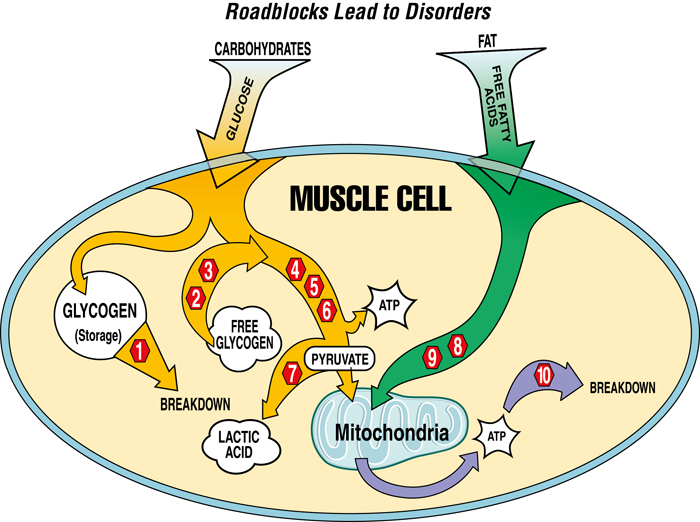
Where the Problems Lie in Each Disease
- Acid maltase deficiency
- Muscle phosphorylase deficiency
- Debrancher enzyme deficiency
- Phosphofructokinase deficiency
- Phosphoglycerate kinase deficiency
- Phosphoglycerate mutase deficiency
- Lactate dehydrogenase deficiency
- Carnitine palymityl transferase deficiency
- Carnitine deficiency
- Myoadenylate deaminase deficiency
Skeletal muscles normally depend on energy from carbohydrates and fats. These fuels can be stored in the muscle (glycogen) or imported directly from the bloodstream (glucose and fatty acids).
When a genetic defect interferes with the processing of specific fuels, energy shortages can occur and toxic byproducts may build up.
Some people may be able to bypass their defects by adjusting diet or exercise to draw energy more efficiently from unaffected pathways.
Looking for more information, support or ways to get involved?
Find MDA
in your Community
-

Search for Clinical Trials
Learn More -

Grants at a Glance
Read More

