
Research Updates Winter 2009
Story includes research items about: centronuclear myopathy; Duchenne, limb-girdle, and Emery-Dreifuss muscular dystrophies; and spinal muscular atrophy.
Blood-vessel narrowing implicated in exercise-related fatigue
Missing or reduced levels of an enzyme known as neuronal nitric oxide synthase (nNOS) at its normal location on muscle-fiber membranes prevents blood vessels that supply active muscles from relaxing normally, leading to exercise-associated fatigue, says a team of researchers from the University of Iowa in Iowa City, the University of Michigan in Ann Arbor, and the University of Washington School of Medicine in Seattle.
 The team, coordinated by longtime MDA grantee Kevin Campbell at the University of Iowa, published its findings online Oct. 26 in Nature. MDA supported Erik Rader at that institution for this work.
The team, coordinated by longtime MDA grantee Kevin Campbell at the University of Iowa, published its findings online Oct. 26 in Nature. MDA supported Erik Rader at that institution for this work.
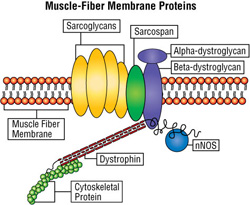
In normal skeletal muscle, nNOS is anchored to the membrane surrounding a muscle fiber via a cluster of proteins (see illustration below). When stimulated by active muscle, nNOS makes a signaling molecule called nitric oxide, which causes a cascade of effects that leads to relaxation of blood vessels and an increase in blood flow to active muscles.
When the protein cluster in the muscle-fiber membrane is disrupted, as occurs in Duchenne muscular dystrophy (DMD) and type 2D limb-girdle muscular dystrophy (LGMD2D), nNOS loses its anchor to the membrane, affecting events downstream of the nitric oxide signaling pathway.
To probe the molecular basis of the exercise-induced fatigue common in people with neuromuscular diseases, the researchers conducted a series of experiments in which they assessed post-exercise activity in mice with and without properly anchored nNOS.
Study results ruled out cardiac dysfunction, inflammation, pain, muscle damage and lack of muscle force as causes of the exercise-related fatigue, and suggested instead that it was caused by a loss of membrane-localized nNOS.
 The team found a similar deficit in nNOS at the muscle-cell membrane in tissue samples from human patients with a wide range of myopathies, suggesting a shared mechanism of fatigue.
The team found a similar deficit in nNOS at the muscle-cell membrane in tissue samples from human patients with a wide range of myopathies, suggesting a shared mechanism of fatigue.
The investigators next either blocked the nNOS signaling pathway or caused vasoconstriction (blood-vessel narrowing) with drugs. Mice treated this way, although healthy, experienced fatigue after mild exercise similar to that experienced by mice with muscular dystrophy or with deficient nNOS. This finding suggests that the absence of properly located nNOS leads to deficient signaling to blood vessels that feed active muscles, thus causing fatigue after only mild exercise.
In further studies, the investigators eliminated the exaggerated fatigue response when they treated mice deficient in nNOS with a phosphodiesterase inhibitor, a drug such as sildenafil (Viagra) that enhances the blood-vessel relaxation signal.
 As indicated in another recent study performed by a research team that included MDA grantee Basil Petrof at McGill University in Montreal, the effect of Viagra on blood vessels extends to the coronary arteries. Those investigators found treatment with sildenafil significantly improved heart function in mice missing the dystrophin protein and showing a disease resembling DMD.
As indicated in another recent study performed by a research team that included MDA grantee Basil Petrof at McGill University in Montreal, the effect of Viagra on blood vessels extends to the coronary arteries. Those investigators found treatment with sildenafil significantly improved heart function in mice missing the dystrophin protein and showing a disease resembling DMD.
Lab-engineered protein increased utrophin production in DMD mice
MDA-supported researchers at the Montreal Neurological Institute of McGill University substantially improved muscle health and resistance to mechanical stress in mice with a disease resembling human Duchenne muscular dystrophy (DMD) by increasing production and distribution of a protein known as utrophin.
The investigators, including MDA grantees George Karpati and Josephine Nalbantoglu, published their findings online Oct. 21 in the Journal of Biological Chemistry.
In their report, they describe how they designed a chemical switch they dubbed zinc-finger protein 51 (ZFP51) that activated utrophin genes, thereby causing muscle fibers to make more utrophin protein and expanding utrophin’s location from one small area to the whole fiber.
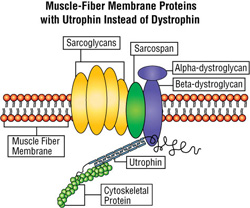
Utrophin is a protein similar in structure to dystrophin, the protein missing in boys with DMD and in DMD mice. Unlike dystrophin, the utrophin gene remains intact in DMD patients and dystrophin-deficient mice, and its protein product has a small protective effect when dystrophin is absent.
 However, utrophin is unable to effectively offset the absence of dystrophin, in part because its structure is slightly different, but mostly because there isn’t enough of it and it’s concentrated in only one place, at the junction of nerve and muscle fibers. In contrast, dystrophin is normally found bordering the entire muscle fiber, and it plays a role in stabilizing the fiber membrane. In addition, protein clusters constructed with utrophin don’t include nNOS. (See "Blood-vessel narrowing" section above.)
However, utrophin is unable to effectively offset the absence of dystrophin, in part because its structure is slightly different, but mostly because there isn’t enough of it and it’s concentrated in only one place, at the junction of nerve and muscle fibers. In contrast, dystrophin is normally found bordering the entire muscle fiber, and it plays a role in stabilizing the fiber membrane. In addition, protein clusters constructed with utrophin don’t include nNOS. (See "Blood-vessel narrowing" section above.)
When dystrophin-deficient mice were given an injection of ZFP51 genes into one leg muscle and an injection without the ZFP51 gene (a “control”) into the corresponding muscle in the opposite leg, only the legs that received the ZFP51 genes showed benefit. The ZFP51-injected muscles functioned better and had less muscle-fiber destruction. They also showed restoration of a normal cluster of proteins at the muscle-fiber membrane and improved resistance to injury from muscle contractions.
The mice showed no obvious toxic effects from the treatment, although the researchers acknowledge that compounds like ZFP51 would have to undergo extensive testing to ensure they didn’t activate genes other than those targeted.
They would also have to ensure that the ZFP genes and the transporters used to deliver them to muscle would not elicit an unwanted immune response from recipients.
“The ZFP is a small protein, and it will probably not be an immune problem,” Karpati said. He also noted that an improved ZFP gene delivery vehicle (“vector”) they plan to use in future experiments “would make the immunogenicity [ability to cause an immune response] of the vector negligible.”
Nalbantoglu, who coordinated the project, and colleagues, say in their report that their studies “highlight the considerable therapeutic potential of artificial ZFPs for treating DMD.”
Asked whether utrophin-containing protein clusters would be deficient in nNOS, she commented that the effects on exercise-induced fatigue occur downstream of nNOS signaling and that the lack of dystrophin could likely be bypassed.
Sarcospan protein unexpectedly saaves MD-affected muscle fibers
A small protein known as sarcospan appears to have much more potential as a treatment for Duchenne muscular dystrophy (DMD) and perhaps other types of muscular dystrophy (MD) than previously believed, say researchers at the University of California-Los Angeles.
Rachelle Crosbie, who has received intermittent MDA funding since the late 1990s, and colleagues, published the sarcospan findings in the Nov. 3 issue of the Journal of Cell Biology. (Crosbie received support from the National Institutes of Health for this project.)
Sarcospan is part of the cluster of proteins at the muscle-fiber membrane that also includes dystrophin, the protein missing in DMD, and the sarcoglycans, which are missing in some types of limb-girdle muscular dystrophy (LGMD) (see illustration above).
Normally, these clusters, closely spaced at intervals around the periphery of muscle fibers, protect the fibers from injury related to mechanical stress. A loss or deficiency of any of the elements of the cluster, such as occurs in DMD and some LGMDs, can destabilize the entire structure, leaving the muscle fiber vulnerable to damage.
Early in life, utrophin, which is very similar to dystrophin, is in these clusters, but it’s replaced by dystrophin as the muscles mature.
Several studies have shown dystrophin-deficient mice with extra utrophin do better than those without it, and various methods to increase utrophin production are being explored as strategies for DMD treatment. (See “Lab-engineered protein” in section above.) Crosbie and colleagues decided instead to explore raising sarcospan levels in muscle fibers.
Unexpectedly, the presence of extra sarcospan (created by giving dystrophin-deficient mice several sarcospan genes) led to the appearance of utrophin-containing protein clusters all around muscle fibers, not just at the specialized nerve-muscle meeting places where they’re normally found. Moreover, the sarcospan-enriched muscles looked healthier than those of the untreated dystrophin-deficient mice, with fewer degenerating fibers.
The investigators say they don’t believe sarcospan increased utrophin production. Instead, they think it stabilized utrophin and allowed it to expand its role and its territory and to substitute for dystrophin.
They note the small size of the sarcospan gene compared to genes for dystrophin or utrophin would make it relatively easy to administer as a gene therapy, and that its widespread presence in muscle tissue would make an unwanted immune response to it unlikely.
Signal-blocking compound markedly improved heart function in EDMD mice
Treatment with a compound that blocks a signaling pathway in the hearts of mice with a disease resembling human Emery-Dreifuss muscular dystrophy (EDMD) resulted in markedly improved cardiac function compared to that seen in untreated EDMD mice, researchers at Columbia University in New York and the Institute of Myology in Paris found.
Howard Worman, a professor and MDA grantee at Columbia, coordinated a group of investigators who published their results online Oct. 16 in Human Molecular Genetics.
Previous work by Worman and colleagues has shown that mutations (flaws) in the lamin A/C gene, which cause EDMD in mice and in humans, lead to abnormal activation of a signaling pathway in the heart called ERK.
In this recent set of experiments, the investigators gave a compound called PD98059, which blocks the ERK signaling pathway, to mice with a lamin A/C gene mutation, while giving other mice with this mutation an inactive substance (placebo) or no treatment at all. They also studied heart function in normal mice.
After eight weeks, the mice that had received injections of PD98059 showed values similar to the normal mice on four measures of heart function. The untreated and the placebo-treated mice, however, showed markedly different values from the normal mice on two of these measures.
In addition to the functional differences, the researchers noted that the nuclei of cardiac muscle cells differed in shape in the different groups of mice.
The normal and PD98059-treated EDMD mice had cardiac muscle cells with well-rounded, oval nuclei, whereas the nuclei in these cells in untreated or placebo-treated EDMD mice had an abnormal, elongated shape.
The lamin A and lamin C proteins are made from the lamin A/C gene’s instructions, and they form part of the envelope that surrounds each cell nucleus (the “nuclear envelope”). 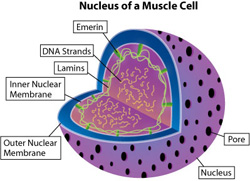 Therefore, these nuclear shape differences could reflect lamin abnormalities.
Therefore, these nuclear shape differences could reflect lamin abnormalities.
EDMD caused by lamin A/C gene mutations is one of two types of EDMD, the other type being caused by mutations in the gene for emerin, another nuclear-envelope protein. The researchers say their findings in a lamin-related EDMD mouse model also may be relevant in emerin-related EDMD, but they have not yet performed any experiments to demonstrate this.
They say their results support the hypothesis that ERK pathway activation caused by lamin abnormalities is an underlying cause of heart-muscle degeneration (cardiomyopathy) in EDMD and that their new results provide evidence that blocking ERK could prevent or delay the onset of heart failure in this disease.
They also note that medications that block ERK have been safely administered to people with cancer.
AAV9 achieves body-wide gene delivery
Researchers at the University of Missouri, the University of North Carolina and Auburn (Ala.) University have found that a vector (a delivery vehicle for transferring genetic material) based on adeno-associated virus 9 (AAV9) reached all the skeletal muscles of young dogs after a single intravenous injection.
The finding is considered significant, because treating muscle disease with gene therapy is likely to require that the therapy reach all muscles after systemic administration.
MDA funded Dongsheng Duan at the University of Missouri, Joe Kornegay at the University of North Carolina, and Bruce Smith at Auburn University for this work.
In a paper published online Sep. 30 in Molecular Therapy, Duan and colleagues describe experiments designed to determine the effectiveness of local and systemic injection of AAV9 as a potential gene therapy delivery method for a host of muscle diseases.
To test the AAV9 delivery system, they first inserted a gene that carries instructions for the alkaline phosphatase (AP) protein into AAV9 shells and injected it directly into muscle.
 Genes injected directly into muscle provoked an unwanted immune reaction when administered to mature animals. However, they were accepted well if given very early in life.
Genes injected directly into muscle provoked an unwanted immune reaction when administered to mature animals. However, they were accepted well if given very early in life.
Researchers next tested whether body-wide gene delivery could be achieved by delivering a high dose or low dose of gene-carrying AAV9 through a vein in each of four animals.
Examinations at two and six weeks after gene transfer showed a dose-dependent response; those that received low-dose AAV9 had fewer AP-positive fibers than the ones that received the high dose.
Six months after the intravenous gene transfer, nearly every skeletal muscle showed efficient AP expression with greater than 50 percent of muscle fibers AP-positive. However, the intravenously administered genes failed to enter the heart muscle, which is affected in many muscle diseases. (Earlier experiments by Duan and colleagues, conducted in mice, found AAV9 targeted the heart.)
“We’re currently working to further improve the technique to achieve not only skeletal muscle but also heart-muscle transduction [entry],” Duan said. “Many muscular dystrophy patients suffer from heart disease. A technique that can reach both skeletal muscle and the heart will be more appealing.”
The investigators note the importance of age at the time of gene delivery and suggest the immune response hurdle may be cleared in newborns, as their immature immune systems may not mount a vigorous response to therapy.
Two gene therapy trials, one in boys with Duchenne muscular dystrophy and another in people with alpha-sarcoglycan-deficient limb-girdle muscular dystrophy (LGMD2D), have received MDA support and are under way. At the same time, however, the search for better delivery vehicles and ways to circumvent an unwanted immune response continues.
Neural stem cells aid survival and function in SMA mice
Mice with a disease resembling human spinal muscular atrophy (SMA) that were treated with specialized stem cells lived 39 percent longer and showed better neuromuscular function than did untreated mice, lending support for development of this type of therapy for SMA.
Investigators coordinated by Giacomo Comi at the University of Milan (Italy), who published their findings in the October issue of the Journal of Clinical Investigation, isolated neural stem cells, which can become motor neurons under certain conditions, from the spinal cords of mouse embryos not affected by SMA.
After exposing these cells to growth-promoting substances and other chemicals and allowing them to multiply in the laboratory, the researchers injected them into the spinal fluid of 1-day-old SMA mice.
The donor cells migrated into the spinal cords of the SMA mice, where they generated cells that appeared to be motor neurons (the muscle-controlling nerve cells that die in SMA). The donor cells may also have aided existing motor neurons in their struggle to survive.
“It is conceivable,” the authors note, “that in the future, stem cell transplantation could be combined with other molecular and pharmacologic approaches to achieve an effective recovery.”
Missing link identified at nerve-muscle junction
MDA grantee Lin Mei recently coordinated a team of scientists who identified a previously missing link necessary to the mechanism that allows signals to pass from nerve to muscle cells. Without these signals, muscles cannot contract.
The protein, known as LRP4, is now known to connect two other proteins, agrin and MuSK, at the neuromuscular junction, the meeting place of nerve and muscle fibers.
“LRP4 is both necessary and sufficient to bind [stick] to agrin and activate MuSK signaling,” note Bin Zhang at the Medical College of Georgia, and colleagues, who published their findings in the Oct. 23 issue of the journal Neuron. The MuSK protein tells the part of the muscle fiber that receives nerve signals to form and maintain clusters of special landing pads for these signals.
Mei also notes that another research group, led by Steven Burden at New York University in Manhattan, independently made the same discovery and published it Oct. 17 in the journal Cell.
Disruptions of signal transmission at the neuromuscular junction can be caused by a mistaken immune-system attack on any of several of its components or by genetic abnormalities in these components.
Immune-system destruction of the signaling protein MuSK or the landing pads, which are called acetylcholine receptors, causes myasthenia gravis (MG), a disease involving interrupted signals that lead to fluctuating weakness.
The investigators note that their results “suggest that LRP4 may be a potential culprit” as well in some diseases of the neuromuscular junction and could become a new target for therapies.
DNA test now available based on MDA grantee's ID of gene for CNM
The University of Chicago Genetics Services Laboratory now offers a DNA test to identify flaws in the dynamin 2 (DNM2) gene that can cause a form of centronuclear myopathy (CNM).
In 2005, MDA grantee Alan Beggs at Children’s Hospital Boston and a multinational team of researchers identified flaws in the DNM2 gene as a cause of CNM. The findings led to this new genetic test.
The lab requires a blood sample, which can be obtained locally. The cost is $2,025, which may or may not be partially covered by insurance. You can arrange for a CNM genetic test through your MDA clinic physician or genetic counselor, who should contact the University of Chicago Genetic Services Laboratories at (773) 834-0555 or ucgslabs@genetics.uchicago.edu.
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information