Friedreich's Ataxia (FRDA)
Friedreich's Ataxia (FRDA)
What is Friedreich's ataxia?
 First described by German physician Nikolaus Friedreich in 1863, Friedreich’s ataxia (known as FA or FRDA) is a neuromuscular disease that mainly affects the nervous system and the heart. FRDA affects about one in 50,000 people worldwide,1 making it the most common in a group of related disorders called hereditary ataxias. It shouldn’t be confused with a group of diseases known as autosomal dominant spinocerebellar ataxias.
First described by German physician Nikolaus Friedreich in 1863, Friedreich’s ataxia (known as FA or FRDA) is a neuromuscular disease that mainly affects the nervous system and the heart. FRDA affects about one in 50,000 people worldwide,1 making it the most common in a group of related disorders called hereditary ataxias. It shouldn’t be confused with a group of diseases known as autosomal dominant spinocerebellar ataxias.
What are the symptoms of FRDA?
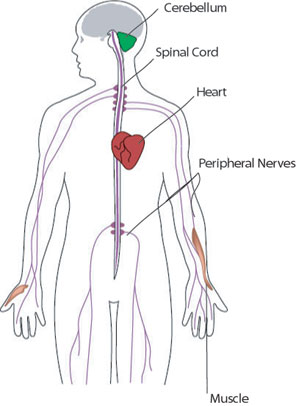
FRDA's major neurological symptoms include muscle weakness and ataxia, a loss of balance and coordination. FRDA mostly affects the spinal cord and the peripheral nerves that connect the spinal cord to the body’s muscles and sensory organs.
FRDA also affects the function of the cerebellum, a structure at the back of the brain that helps plan and coordinate movements. (It doesn’t affect the parts of the brain involved in mental functions, however.)
FRDA's effects on the heart range from mild abnormalities to life-threatening problems in the heart’s musculature. For more, see Signs and Symptoms.
What causes FRDA?
FRDA is a hereditary disease, caused by a defect (mutation) in a gene that can be passed down through a family. Mutations in the gene that carries instructions for a protein called frataxin result in diminished energy production in cells, including those of the nervous system and heart. For more, see Causes/Inheritance.
What is the progression of FRDA?
Onset is typically between 5 and 15 years of age, but FRDA has been diagnosed in people from ages 2 to 50. FRDA progresses slowly, and the sequence and severity of its progression is highly variable. Although there's no cure for FRDA as yet, treatments exist for cardiac symptoms, and there are ways to manage ataxia and muscle weakness. Many people with FRDA lead active lives, going to college, holding careers, getting married and starting families.
What is the status of FRDA research?
Scientists are making definite progress toward better treatments for FRDA. For more about current research in FRDA, see Research.
MDA educational resources
Research
- Friedreich Ataxia Fact Sheet | National Institute of Neurological Disorders and Stroke. Published April 2022. Accessed June 6, 2022. https://www.ninds.nih.gov/health-information/patient-caregiver-education...
Looking for more information, support or ways to get involved?
Find MDA
in your Community
-

Grants at a Glance
Read More -

Search for Clinical Trials
Learn More

