
Taking Aim at Congenital Muscular Dystrophies

Insights from pediatric neurologist Carsten Bönnemann
Update (March 24, 2015): A phase 1 trial of omigapil in children and adolescents 5-16 years old who have merosin-deficient congenital muscular dystrophy and meet other study criteria is open in Bethesda, Md. See Assessment of Safety and Tolerability of Omigapil (CALLISTO), or enter NCT01805024 in the search box at ClinicalTrials.gov.
Not long ago, a diagnosis of congenital muscular dystrophy (CMD) in a baby could not provide much information or comfort for a parent.
It generally meant the child was very weak — often described as “floppy” — at birth, and may have had difficulty breathing or swallowing. The diagnosis, as well as the prognosis, was uncertain, and treatment was almost nonexistent.
 Today, though, there is hope. Supportive care, such as assisted ventilation, has increased survival time, and new knowledge has brought the prospects for disease-altering treatment closer than at any time in history.
Today, though, there is hope. Supportive care, such as assisted ventilation, has increased survival time, and new knowledge has brought the prospects for disease-altering treatment closer than at any time in history.
We recently talked with pediatric neurologist Carsten Bönnemann, a senior investigator at the National Institutes of Health (NIH) in Bethesda, Md., who has had a special interest in the CMDs. Here’s what he had to say:
Q: How is congenital muscular dystrophy defined?
A: One part of the definition of congenital muscular dystrophy is that the age of onset of clinical symptoms is birth or early infancy, with weakness being evident at birth or recognized in infancy.
The second aspect of the definition is that a muscle biopsy shows some evidence of muscular dystrophy — or at least is not inconsistent with it — and does not show findings of any other disorder that has congenital onset. Those aspects of CMD are the cornerstones.
Sometimes, but not always, children with CMD have brain or eye abnormalities; and sometimes — again, not always — their joints are stuck in a fixed position. [A variation on this is found in Ullrich CMD, where some joints are actually more flexible than normal, while others are stiffer.]
Q: What do we now know that we didn’t know a few years ago?
A: On the molecular level, some themes have emerged. If you look at the different forms of congenital muscular dystrophy, most of them fall into two big groups. One group — and it’s by far the majority of patients — is related to defects in proteins that make up the extracellular matrix of muscle tissue or affect proteins that interact with this matrix. [The extracellular matrix is the substance that surrounds the cells of a tissue, such as muscle, providing physical and biochemical support.]
An important role of the matrix around muscle fibers is force transmission. For a muscle to pull against bones, it needs to have contact with something that transmits force from muscle across the membrane into the matrix and onto the tendons and bones.
And when all is going as it should, the matrix is a transmitter not only of force, but also of chemicals, such as growth factors, that muscles need to stay healthy.
So the matrix is a physical and biochemical supporting structure for survival and regeneration of muscle. You need the matrix to be able to regenerate muscle. When cells lose touch with the surrounding matrix, trouble follows.
Tissues have evolved to take care of this problem, albeit imperfectly, by causing cells that lose their matrix connections to self-destruct, or “commit suicide.” [This self-destruction process is known as apoptosis.]
If a cell loses connection to the matrix, it’s prone to shutting itself off. And that seems to be part of the story with muscle fibers. If the matrix connection is abnormal or different, there seems to be a higher propensity for the fibers to undergo apoptosis, or at least not stay healthy.
In addition, when the matrix senses something abnormal in its midst, it sometimes triggers inflammatory chemicals that attack the muscle fiber. Eventually, if the inflammation is strong enough, the inflamed tissue turns into a kind of hardened scar. [This process is called fibrosis.]
Presumably, both apoptosis and fibrosis evolved to defend muscle and other tissues from invading microbes, out-of-control cancer cells and other threats, and to support repair and regeneration following an acute injury. Unfortunately, though, as with so many defense mechanisms, they have their downside: When an abnormal protein initiates damage to a tissue on a permanent basis, as is the case in a genetic disorder, the defense can turn on but not turn off again and thereby further damage the tissue.
A second group of CMDs — a much smaller group — has roots in proteins that have their main effect inside muscle fibers, rather than in the matrix-fiber interaction. [For instance, some of these proteins affect how muscle fibers receive signals from the nervous system and how they handle calcium.]
It’s now known that at least 15 genes [the “blueprints” for proteins] can, when flawed, lead to various forms of CMD. And with that knowledge has come much more understanding of what goes wrong at the cellular level in these diseases.
(See CMD: A Cellular View below to learn more about the muscle-fiber membrane and matrix.)
Q: Has the recently gained knowledge about specific CMD-related genes and proteins been put to use in development of therapies?
A: Much of what’s now known about the congenital muscular dystrophies can, fortunately, be used to develop therapies.
There are gene-specific approaches, and then there are the more global approaches. A gene-specific approach would be gene replacement therapy. That would apply to some of the conditions we’ve been discussing more than others.
Although not yet being tested in humans, gene therapy studies using some of the CMD genes, such as the fukutin-related proteingene and the LARGE gene, are under way in laboratory mice.
Q: What drugs are being developed to address cell death, inflammation and fibrosis in CMD?
 A: There are some less specific, more global approaches for treating CMD that target two of the defensive-turned destructive mechanisms that occur when cells lose contact with their matrix: apoptosis, or cell death, and inflammation.
A: There are some less specific, more global approaches for treating CMD that target two of the defensive-turned destructive mechanisms that occur when cells lose contact with their matrix: apoptosis, or cell death, and inflammation.
We think apoptosis is a result of the disengagement of muscle from its matrix. We don’t know how far downstream of the genetic mutation this happens, but it seems to be present, particularly in muscles deficient in laminin 211, which is also known asmerosin. It’s been less studied in other CMD types, but it’s probably present in the Ullrich type as well. [Ullrich CMD is caused by mutations in genes for collagen 6, a matrix protein.]
An experimental drug called omigapil and an existing drug called doxycycline appear to combat apoptosis and have shown some promise in treating mice with a deficiency of merosin, or laminin 211. These mice have a disorder that closely resembles human merosin-deficient CMD, prompting investigators to propose trying them in patients.
Another existing drug that seems to have some anti-apoptotic properties iscyclosporine, and it’s been tried in a mouse model that has a mutation in a collagen 6 gene, where it had some positive effects on muscle structure. That mouse is not a great mimic of Ullrich CMD, but it does show some apoptosis, and it leads me to believe that targeting this form of CMD with an anti-apoptotic drug might be worth a try.
In fact, in 2011, a very small study in children with Ullrich CMD conducted in Italy suggested cyclosporine might have a positive effect on the apoptosis seen in muscle. However, the drug has many toxic side effects, such as vulnerability to serious infections and dysfunction of the kidneys and liver, and is not an ideal candidate for long-term treatment of CMD.
Targeting inflammation and its ultimate consequence — fibrosis — is also high on the list of possible interventions for CMD. When the matrix goes into chronic fibrosis mode, that’s detrimental to muscle regeneration.
An existing drug that seems to impede that process and improve the disease course in merosin-deficient mice is losartan. And the anti-apoptotic drug doxycycline also has anti-inflammatory properties.
It’s a very reasonable target to try to inhibit fibrosis as a disease driver. You’re not treating the primary disease. But you’re treating a downstream, secondary driver of the disease, so you would expect to ameliorate, but not cure, it. Losartan has been tried in the merosin-deficient mouse in two different labs, with very similar, beneficial results.
Corticosteroid drugs like prednisone [which are anti-inflammatory] have on occasion been given to patients with some types of CMD, and a clinical trial to test prednisone is being considered.
Q: What is being done to speed up CMD clinical trials or the drug development process?
A: Getting ready for CMD clinical trials is exciting, but it’s also tricky. A crucial first step is to study the natural course of each of the subtypes of the disease, so that investigators can see whether or not a particular medication being tested changes it. [This process is known as defining outcome measures]. Just to define the motor outcome measures, and how they change over a period of time, is challenging in this group of patients, but you have to do that to be in a position to do clinical trials.
Q: Are there any steps people can take right now to help advance CMD research?
A: Along with Anne Rutkowski, a physician and the mother of a child with CMD, as well as others, we’ve started an international registry [database] to gather information about people with the various subtypes of CMD.
Known as the Congenital Muscle Disease International Registry, it’s under he auspices of Cure CMD. The questionnaire is designed to be filled out online but also can be completed on paper. It includes inquiries about demographics, disease specifics and diagnosis. This is an essential step on the way to clinical trials in the CMDs.
Q: What’s the take-home message for CMD-affected families today?
A: By understanding these conditions better, by establishing a definitive diagnosis in affected children and adults, and by having a much better grasp on medical interventions, such as breathing support, we already are able to make a big difference in the life of a person with CMD.
Now that we also understand more about the individual and common causes of these conditions, we can develop better treatment approaches in the lab, and at the same time, we’re putting in place the pieces necessary for meaningful clinical trials in the CMDs.
We’re connecting the dots toward the development of real therapies, but it’s going to take the efforts of affected people and their families, scientists, physicians and other health professionals, as well as funding agencies, regulators and industry.
Congenital Muscular Dystrophy Care Guidelines
In 2011, the first-ever CMD care guidelines were released, written in family friendly language. To view them, go to MDA Congenital Muscular Dystrophy Care Guidelines, or ask your local MDA office for a printout.
CMD: The Cellular View
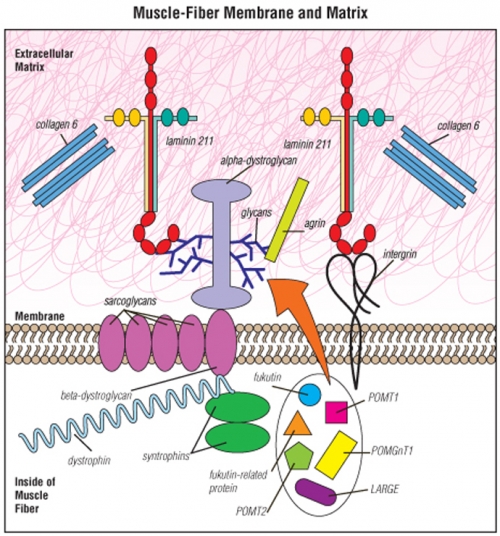 Muscle fibers are surrounded by a membrane that separates the inside of the fiber from the material outside the fiber — the extracellular matrix.
Muscle fibers are surrounded by a membrane that separates the inside of the fiber from the material outside the fiber — the extracellular matrix.
Most of the molecular defects that cause congenital muscular dystrophies affect proteins in the extracellular matrix, such as laminin 211, integrin, collagen 6 or alpha-dystroglycan.
The proteins known as fukutin, fukutin-related protein, POMT1, POMT2, POMGnT1, LARGE and others all participate in a special “sugar coating” (glycosylation) of alpha-dystroglycan. The sugar coating is shown as the blue, branchlike structures in this illustration. The branches are known as glycans.
Various other proteins shown — such as the sarcoglycansand dystrophin — can, when flawed or missing, cause muscular dystrophies other than CMD.
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information